Kisspeptin-10, human TFA 是一种有效的血管收缩剂和血管生成抑制剂。Kisspeptin-10, human TFA 通过其受体 GPR54 来抑制肿瘤转移作用。Kisspeptin-10-GPR54 系统在胚胎肾脏发育中起重要作用。Kisspeptin-10/GPR54 信号通过 NFATc4 介导的 BMP2 表达来诱导成骨细胞分化。
编号:139448
CAS号:374675-21-5
单字母:H2N-YNWNSFGLRF-NH2
| 编号: | 139448 |
| 中文名称: | 亲吻促动素-10(人源)、Kisspeptin-10, Metastin(45-54), Human |
| 英文名: | Kisspeptin-10Metastin(45-54), Human |
| CAS号: | 374675-21-5 |
| 单字母: | H2N-YNWNSFGLRF-NH2 |
| 三字母: | H2N N端氨基 -Tyr酪氨酸 -Asn天冬酰胺 -Trp色氨酸 -Asn天冬酰胺 -Ser丝氨酸 -Phe苯丙氨酸 -Gly甘氨酸 -Leu亮氨酸 -Arg精氨酸 -Phe苯丙氨酸 -NH2C端酰胺化 |
| 氨基酸个数: | 10 |
| 分子式: | C63H83N17O14 |
| 平均分子量: | 1302.44 |
| 精确分子量: | 1301.63 |
| 等电点(PI): | - |
| pH=7.0时的净电荷数: | 2.97 |
| 酸性基团个数: | 2 |
| 碱性基团个数: | 疏水 |
| 平均亲水性: | -1.1 |
| 疏水性值: | -0.51 |
| 外观与性状: | 白色粉末状固体 |
| 消光系数: | 6990 |
| 来源: | 人工化学合成,仅限科学研究使用,不得用于人体。 |
| 纯度: | 95%、98% |
| 盐体系: | 可选TFA、HAc、HCl或其它 |
| 储存条件: | 负80℃至负20℃ |
| 标签: | 抑制剂相关肽(Inhibitor Peptide) 现货多肽 血管紧张素(Angiotensin) 吻素(Kisspeptin) |
Kisspeptin-10, human 是一种有效的血管收缩剂和血管生成抑制剂。Kisspeptin-10, human 通过其受体 GPR54 来抑制肿瘤转移作用。Kisspeptin-10-GPR54 系统在胚胎肾脏发育中起重要作用。Kisspeptin-10/GPR54 信号通过 NFATc4 介导的 BMP2 表达来诱导成骨细胞分化。
Kisspeptin-10, human is a potent vasoconstrictor and inhibitor of angiogenesis. Kisspeptin-10, human acts as a tumor metastasis suppressor via its receptor GPR54. Kisspeptin-10-GPR54 system plays an important role in embryonic kidney development. Kisspeptin-10/GPR54 signaling induces osteoblast differentiation via NFATc4-mediated BMP2 expression[1].
Kisspeptin-10, human TFA is a potent vasoconstrictor and inhibitor of angiogenesis, it acts as a tumor metastasis suppressor via its receptor GPR54. Kisspeptin-10/GPR54 signaling induces osteoblast differentiation via NFATc4-mediated BMP2 expression. Kisspeptin-10 (KP-10) and its receptor GPR54 are key components in the regulation of GnRH secretion in humans and other mammals. Kisspeptin-10 protein binds to GPR54. Activation of Kisspeptin-10 suppresses pulmonary human melanoma and Kisspeptin-10 is a metastasis suppressor in breast cancer cells.
In vivo, intravenous infusion of kisspeptin-10 (7.5, 35, and 100 nM) induces a dose-dependent increase in LH secretion. The stimulatory effect of kisspeptin-10 (100 n nM) on LH secretion is blocked by the GnRH antagonist cetrorelix, precluding a singular action on gonadotropes. Kisspeptin-10 inhibits angiogenesis in vivo. Kp-10 inhibits tumor growth in SCID mice xenografted with human prostate cancer cells (PC-3) through inhibiting tumor angiogenesis.
定义
酶是用于生化反应的非常有效的催化剂。它们通过提供较低活化能的替代反应途径来加快反应速度。酶作用于底物并产生产物。一些物质降低或什至停止酶的催化活性被称为抑制剂。
发现
1965年,Umezawa H分析了微生物产生的酶抑制剂,并分离出了抑制亮肽素和抗痛药的胰蛋白酶和木瓜蛋白酶,乳糜蛋白酶抑制的胰凝乳蛋白酶,胃蛋白酶抑制素抑制胃蛋白酶,泛磷酰胺抑制唾液酸酶,乌藤酮抑制酪氨酸羟化酶,多巴汀抑制多巴胺3-羟硫基嘧啶和多巴胺3-羟色胺酶酪氨酸羟化酶和多巴胺J3-羟化酶。最近,一种替代方法已应用于预测新的抑制剂:合理的药物设计使用酶活性位点的三维结构来预测哪些分子可能是抑制剂1。已经开发了用于识别酶抑制剂的基于计算机的方法,例如分子力学和分子对接。
结构特征
已经确定了许多抑制剂的晶体结构。已经确定了三种与凝血酶复合的高效且选择性的低分子量刚性肽醛醛抑制剂的晶体结构。这三种抑制剂全部在P3位置具有一个新的内酰胺部分,而对胰蛋白酶选择性最高的两种抑制剂在P1位置具有一个与S1特异性位点结合的胍基哌啶基。凝血酶的抑制动力学从慢到快变化,而对于胰蛋白酶,抑制的动力学在所有情况下都快。根据两步机理2中稳定过渡态络合物的缓慢形成来检验动力学。
埃米尔•菲舍尔(Emil Fischer)在1894年提出,酶和底物都具有特定的互补几何形状,彼此恰好契合。这称为“锁和钥匙”模型3。丹尼尔·科什兰(Daniel Koshland)提出了诱导拟合模型,其中底物和酶是相当灵活的结构,当底物与酶4相互作用时,活性位点通过与底物的相互作用不断重塑。
在众多生物活性肽的成熟过程中,需要由其谷氨酰胺(或谷氨酰胺)前体形成N末端焦谷氨酸(pGlu)。游离形式并与底物和三种咪唑衍生抑制剂结合的人QC的结构揭示了类似于两个锌外肽酶的α/β支架,但有多个插入和缺失,特别是在活性位点区域。几种活性位点突变酶的结构分析为针对QC相关疾病5的抑制剂的合理设计提供了结构基础。
作用方式
酶是催化化学反应的蛋白质。酶与底物相互作用并将其转化为产物。抑制剂的结合可以阻止底物进入酶的活性位点和/或阻止酶催化其反应。抑制剂的种类繁多,包括:非特异性,不可逆,可逆-竞争性和非竞争性。可逆抑制剂 以非共价相互作用(例如疏水相互作用,氢键和离子键)与酶结合。非特异性抑制方法包括最终使酶的蛋白质部分变性并因此不可逆的任何物理或化学变化。特定抑制剂 对单一酶发挥作用。大多数毒药通过特异性抑制酶发挥作用。竞争性抑制剂是任何与底物的化学结构和分子几何结构非常相似的化合物。抑制剂可以在活性位点与酶相互作用,但是没有反应发生。非竞争性抑制剂是与酶相互作用但通常不在活性位点相互作用的物质。非竞争性抑制剂的净作用是改变酶的形状,从而改变活性位点,从而使底物不再能与酶相互作用而产生反应。非竞争性抑制剂通常是可逆的。不可逆抑制剂与酶形成牢固的共价键。这些抑制剂可以在活性位点附近或附近起作用。
功能
工业应用中, 酶在商业上被广泛使用,例如在洗涤剂,食品和酿造工业中。蛋白酶用于“生物”洗衣粉中,以加速蛋白质在诸如血液和鸡蛋等污渍中的分解。商业上使用酶的问题包括:它们是水溶性的,这使得它们难以回收,并且一些产物可以抑制酶的活性(反馈抑制)。
药物分子,许多药物分子都是酶抑制剂,药用酶抑制剂通常以其特异性和效力为特征。高度的特异性和效力表明该药物具有较少的副作用和较低的毒性。酶抑制剂在自然界中发现,并且也作为药理学和生物化学的一部分进行设计和生产6。
天然毒物 通常是酶抑制剂,已进化为保护植物或动物免受天敌的侵害。这些天然毒素包括一些已知最剧毒的化合物。
神经气体( 例如二异丙基氟磷酸酯(DFP))通过与丝氨酸的羟基反应生成酯,从而抑制了乙酰胆碱酯酶的活性位点。
参考
1、Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
2、Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
3、Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
4、Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
5、Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
6、Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
Definition
Enzymes are very efficient catalysts for biochemical reactions. They speed up reactions by providing an alternative reaction pathway of lower activation energy. Enzyme acts on substrate and gives rise to a product. Some substances reduce or even stop the catalytic activities of enzymes are called inhibitors.
Discovery
In 1965, Umezawa H analysed enzyme inhibitors produced by microorganisms and isolated leupeptin and antipain inhibiting trypsin and papain, chymostatin inhibiting chymotrypsin, pepstatin inhibiting pepsin, panosialin inhibiting sialidases, oudenone inhibiting tyrosine hydroxylase, dopastin inhibiting dopamine 3-hydroxylase, aquayamycin and chrothiomycin inhibiting tyrosine hydroxylase and dopamine J3-hydroxylase . Recently, an alternative approach has been applied to predict new inhibitors: rational drug design uses the three-dimensional structure of an enzyme's active site to predict which molecules might be inhibitors 1. Computer-based methods for identifying inhibitor for an enzyme have been developed, such as molecular mechanics and molecular docking.
Structural Characteristics
The crystal structures of many inhibitors have been determined. The crystal structures of three highly potent and selective low-molecular weight rigid peptidyl aldehyde inhibitors complexed with thrombin have been determined. All the three inhibitors have a novel lactam moiety at the P3 position, while the two with greatest trypsin selectivity have a guanidinopiperidyl group at the P1 position that binds in the S1 specificity site. The kinetics of inhibition vary from slow to fast with thrombin and are fast in all cases with trypsin. The kinetics are examined in terms of the slow formation of a stable transition-state complex in a two-step mechanism 2.
Emil Fischer in 1894 suggested that both the enzyme and the substrate possess specific complementary geometric shapes that fit exactly into one another.This is known as "the lock and key" model 3. Daniel Koshland suggested induced fit model where substrate and enzymes are rather flexible structures, the active site is continually reshaped by interactions with the substrate as the substrate interacts with the enzyme 4.
N-terminal pyroglutamate (pGlu) formation from its glutaminyl (or glutamyl) precursor is required in the maturation of numerous bioactive peptides. The structure of human QC in free form and bound to a substrate and three imidazole-derived inhibitors reveals an alpha/beta scaffold akin to that of two-zinc exopeptidases but with several insertions and deletions, particularly in the active-site region. The structural analyses of several active-site-mutant enzymes provide a structural basis for the rational design of inhibitors against QC-associated disorders 5.
Mode of Action
Enzymes are proteins that catalyze chemical reactions. Enzymes interact with substrate and convert them into products. Inhibitor binding can stop a substrate from entering the enzyme's active site and/or hinder the enzyme from catalyzing its reaction. There are a variety of types of inhibitors including: nonspecific, irreversible, reversible - competitive and noncompetitive. Reversible inhibitors bind to enzymes with non-covalent interactions like hydrophobic interactions, hydrogen bonds, and ionic bonds. Non-specific methods of inhibition include any physical or chemical changes which ultimately denature the protein portion of the enzyme and are therefore irreversible. Specific Inhibitors exert their effects upon a single enzyme. Most poisons work by specific inhibition of enzymes. A competitive inhibitor is any compound which closely resembles the chemical structure and molecular geometry of the substrate. The inhibitor may interact with the enzyme at the active site, but no reaction takes place. A noncompetitive inhibitor is a substance that interacts with the enzyme, but usually not at the active site. The net effect of a non competitive inhibitor is to change the shape of the enzyme and thus the active site, so that the substrate can no longer interact with the enzyme to give a reaction. Non competitive inhibitors are usually reversible. Irreversible Inhibitors form strong covalent bonds with an enzyme. These inhibitors may act at, near, or remote from the active site .
Functions
Industrial application, enzymes are widely used commercially, for example in the detergent, food and brewing industries. Protease enzymes are used in 'biological' washing powders to speed up the breakdown of proteins in stains like blood and egg. Problems using enzymes commercially include: they are water soluble which makes them hard to recover and some products can inhibit the enzyme activity (feedback inhibition) .
Drug molecules, many drug molecules are enzyme inhibitors and a medicinal enzyme inhibitor is usually characterized by its specificity and its potency. A high specificity and potency suggests that a drug will have fewer side effects and less toxic. Enzyme inhibitors are found in nature and are also designed and produced as part of pharmacology and biochemistry 6.
Natural poisons are often enzyme inhibitors that have evolved to defend a plant or animal against predators. These natural toxins include some of the most poisonous compounds known.
Nerve gases such as diisopropylfluorophosphate (DFP) inhibit the active site of acetylcholine esterase by reacting with the hydroxyl group of serine to make an ester.
References
Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
定义
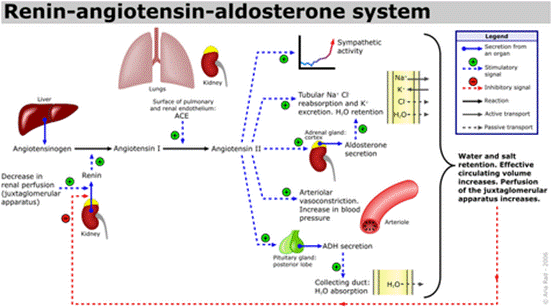
血管生成素(ANG),一种14,124 Da的蛋白质,与肿瘤进展中的血管生成有关。它由肿瘤细胞分泌,肿瘤细胞是新生血管形成的有效诱导剂1。同时血管生成素可引起血管收缩并促使血压升高。它是肾素-血管紧张素系统的一部分,这是降低血压的药物的主要靶标。血管紧张素(ANG)是血管紧张素原(AGT)在肾素和一系列酶的作用下形成的一组寡肽激素。
血管紧张素是一种肽类激素,引起血管收缩及随后血压升高。血管紧张素是一种寡肽类激素,具有强大的致渴作用。它是由前体分子血管紧张素原生成的,血管紧张素原是肝脏中产生的血清球蛋白。血管紧张素在肾素-血管紧张素系统中起重要作用(1)。肾素作用于血管紧张素原,产生血管紧张素I。在肾小球旁细胞中,当肾交感神经兴奋、肾内血压降低或运送到致密斑的Na+和Cl-离子降低,致密斑感受到较少的Na+时,肾小球旁细胞释放的肾素增加。肾素裂解血管紧张素原中亮氨酸(Leu)和缬氨酸(Val)之间的肽键,生成含有十个氨基酸的多肽(DES-ASP),即血管紧张素I,血管紧张素I似乎没有任何的生物活性,仅仅是作为血管紧张素II的前体。
发现
1885年,Fett等人首先从由HT-29人结肠腺癌细胞1调节的培养基中分离血管生成素,然后从正常哺乳动物的血浆2和牛奶中分离出血管生成素。随后已从人,牛,兔,猪和小鼠的血清和牛乳中分离出它。血管生成素具有核糖核酸分解活性,与胰腺RNAse A 具有33%的序列同源性。合成肽'H-Glu-Asn-Gly-Leu-Pro-Val-His-Leu-Asp-Gln-Ser-Ile-Phe-Arg-Arg-OH(108-122)对应于ANG抑制血管生成素的酶和生物活性。几种C末端合成肽,包括(Ang 108-123),可显着降低血管生成素诱导的新血管形成。
结构特点
Acharya等人在2.4 0 A时确定了人类抗原的晶体结构。总体结构具有肾形三级褶皱,让人联想到RNaseA。核糖核酸裂解活性中心(His-13,His-114和Lys-40)和假定的受体结合位点,这两个关键参与生物学功能是不同于Rnase A 。 分子的中心核心由带有一对反平行扭曲的P结构组成,形成了主拓扑,顶点处有残基Ser-72和Gly-99。在这些中央链的任一侧的两个附加链(残基41-47; 111-116)完成了主要的片状结构。结构中存在3个螺旋H1,残基3-14,H2,残基22-33和H3,残基49-58。
作用机理
ANG四个方面已经发现,是必要的ANG诱导的血管生成的过程中,ANG发挥其核糖核酸活动- ANG有一个非常弱的10 5 - 10 6比核糖核酸酶答:这更低的核糖核酸活动,因为嘧啶结合谷氨酰胺(Gln)117残基“阻碍”了ANG的位点。然而,ANG的核糖核酸分解活性对于血管生成至关重要。ANG提神基底膜降解- ANG结合到一个上肌动蛋白内皮细胞表面 ANG-肌动蛋白复合物从细胞表面解离并加速组织型纤溶酶原激活物(tPA)催化的纤溶酶从纤溶酶原的生成。ANG-肌动蛋白复合物促进基底膜和细胞外基质的降解。该复合物允许内皮细胞渗透并迁移到血管周组织中。基底膜降解是血管生成的基本特征。ANG激活信号转导- ANG结合至170-kDa的 位于内皮细胞表面上的受体和引发第二信使系统。ANG与细胞表面肌动蛋白的结合导致细胞相关蛋白酶系统的活化,从而促进细胞侵袭。ERK1 / 2,蛋白激酶B / Akt1 已经提出通过ANG刺激来激活这些途径。ANG核易位-血管生成素通过受体介导的内吞作用和核定位序列辅助的核输入在内皮细胞中进行核易位。核定位信号(NLS)位于蛋白质的31-RRRGL-35中。核易位后,它增强rRNA转录。
功能
血管紧张素受体存在于许多组织类型中,包括肾上腺皮质,肾小球,心脏,下丘脑,肝脏,胰腺,垂体,血小板,肾小管,子宫和血管平滑肌。通过放射性配体与拮抗剂的结合已鉴定出两种高亲和力受体亚型:氯沙坦(DuP 753 / MK954)鉴定AT1受体;PD123177和CGP42112A是AT2受体的标记。血管紧张素II可能在体液系统外的组织中局部产生。例如,它存在于大脑,肾脏和心脏。在大脑中,七肽血管紧张素(1-7)模仿血管紧张素II的某些作用,但可能直接由血管紧张素I形成。心脏中有非ACE介导的血管紧张素II产生的证据。血管内血管紧张素II受体与血管加压素和其他垂体后叶激素的中枢释放,交感神经外流增加,口渴反应以及可能的认知功能有关。血管紧张素II对心脏以及对生长/肥大的正性和变时性作用; 控制醛固酮的释放以及皮质醇和醛固酮分泌之间的平衡;并调节钠,氯和碳酸氢盐在肾脏内的运输。
ANG在血管生成中的功能-作为关键的血管生成因子,ANG与内皮细胞和平滑肌细胞相互作用,诱导多种细胞应答,包括细胞迁移,侵袭,增殖和肾小管结构形成。还已经报道ANG可直接诱导癌细胞的增殖。最近,ANG 基因被确定为潜在的肌萎缩性侧索硬化症(ALS)相关基因6。ANG可诱导多种人类癌症中的肿瘤生长,包括乳腺癌,宫颈癌,结肠癌,结肠直肠癌,子宫内膜癌,胃癌,肝癌,肾癌,卵巢癌,胰腺癌,前列腺癌和尿路上皮癌,以及星形细胞瘤,白血病,淋巴瘤,黑素瘤,骨肉瘤,和肾母细胞“肿瘤 6。ANG可能与肌萎缩性侧索硬化症有关-肌萎缩性侧索硬化症(ALS)是一种进展性迟发性神经退行性疾病,会影响上下运动神经元(MNs)。血管内皮生长因子是第一个被证明与ALS 7发病有关的血管生成因子。
Definition
The KISS1 receptor (previously designated GPR54) has been paired with biologically active cleavage peptides of the KiSS-1 gene product, the kisspeptins (KP).
Discovery
KP was originally identified in 1996 from a metastasis suppressor gene, KiSS-1, in malignant melanomas. Initially, the largest cleavage product, KP-54, was identified for its ability to suppress metastatic potential in human melanoma cells. Its expression also resulted in suppression of melanoma metastasis in athymic nude mice and it was therefore termed metastin1. In 2001, one of the orphans KISS1 (previously designated GPR54, AXOR12, hOT7T175) was paired with three biologically active cleavage peptides of the kisspeptin gene (KiSS-1) gene product isolated from human placenta, the KP’s (KP)-54, KP-13 and KP-10 2, 3. The KP, as a collective group, where individual KP is referred to, their amino acid sequence length e.g., KP-54, KP-13 and KP-10.
Structural Characteristics
The full-length KP protein (KP-145) has a PEST sequence (proline, glutamic acid, serine, threonine and aspartic acid residue-rich sequence). This motif predisposes proteins for ubiquitination and proteosome degradation and suggests that cytosolic KP-145 would have a short half-life 4. In rat and mouse, the longest KP cleavage fragment is composed of 52 amino acids. Although overall homology of human KiSS-1 gene products to rat and mouse is relatively low (B52%), KP-10 is highly conserved between human, mouse and rat, with only one amino acid difference in the sequence between species 1.
Mode of Action
Activation of KISS1 (GPR54) results in intracellular calcium mobilization that is not affected by pertussis toxin and does not result in changes in cAMP accumulation, suggesting that it is a Gq-coupled receptor. KP activation of KISS1 (GPR54) has been shown to simultaneously result in release of arachidonic acid and stimulation of the mitogen-activated protein kinase (MAPKs) extracellular signal-regulated kinase (ERK) 1 and ERK2. This has been attributed to increased phosphorylation of MAPK 5
Functions
KP and matrix metalloproteinases- Downregulation of one or both of the gelatinase matrix metalloproteinases (MMPs), MMP-2 and MMP-9, by KP has been shown. KP has been described as regulator of MMPs at both the transcriptional and protein level. Transcriptional changes have been shown not to function through the MAPK signalling pathways. Instead, nuclear factor-kB binding to the MMP-9 promoter region, necessary for expression of MMP-9 6.
KP and cancer metastasis- Direct interaction of KiSS-1 has been identified with two transcription factors, activator protein-2a and specificity protein-1, both of which have been shown to be important regulators of genes involved in tumourigenesis, metastasis and development 7.
KP and placentation- Microarray analysis confirmed the higher KISS1 (GPR54) expression in first trimester invasive trophoblasts, when compared to non/low-invasive term cells.
KISS1 (GPR54) – an unexpected molecular switch for puberty- In 2003, three different groups identified KISS1 (GPR54) as an unexpected molecular switch for puberty 8.
KP and GnRH release-The signalling pathway showed that injection of KP-10 and KP-54 directly into the lateral cerebral ventricle of the mouse brain potently stimulated LH and follicle-stimulating hormone (FSH) secretion, an effect that could be blocked by pretreatment with the GnRH antagonist acyline 8.
KP neurones in the brain- Approximately 75% of GnRH neurones co-express KISS1 (GPR54), a finding that has been confirmed in sheep, where intra cerebral injection of KP resulted in direct release of GnRH into the cerebrospinal fluid 8.
Direct effects of KP on the testes-Continuous chronic administration of KP in male rats resulted in decreased testicular weight and degeneration of the seminiferous tubules, leading to the hypothesis that KP may alter testicular blood flow 8.
References
1. Lee JH, Miele ME, Hicks DJ, Phillips KK, Trent JM, Weissman BE, Welch DR. (1996). KiSS-1, a novel human malignant melanoma metastasissuppressor gene. J Natl Cancer Inst., 88: 1731-1737.
2. Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brézillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, Blanpain C, Schiffmann SN, Vassart G, Parmentier M. (2001). The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem., 276(37):34631-34636.
3. Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, Ishibashi Y, Watanabe T, Asada M, Yamada T, Suenaga M, Kitada C, Usuki S, Kurokawa T, Onda H, Nishimura O, Fujino M. (2001). Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature., 411 (6837): 613–617.
4. Harms JF, Welch DR, Miele ME (2003). KISS1 metastasis suppression and emergent pathways. Clin Exp Metastasis., 20(1):11-18.
5. Becker JA, Mirjolet JF, Bernard J, Burgeon E, Simons MJ, Vassart G, Parmentier M, Libert F. (2005). Activation of GPR54 promotes cell cycle arrest and apoptosis of human tumor cells through a specific transcriptional program not shared by other Gq-coupled receptors. Biochem Biophys Res Commun., 326(3):677-686.
6. Qiao C, Wang CH, Shang T, Lin QD (2005). Clinical significance of KiSS-1 and matrix metalloproteinase-9 expression in trophoblasts of women with preeclampsia and their relation to perinatal outcome of neonates. Zhonghua Fu Chan Ke Za Zhi., 40(9): 585–590.
7. Mitchell DC, Abdelrahim M, Weng J, Stafford LJ, Safe S, Bar-Eli M, Liu M.(2006). Regulation of KiSS-1 metastasis suppressor gene expression in breast cancer cells by direct interaction of transcription factors activator protein-2alpha and specificity protein-1. J Biol Chem., 281: 51–58.
8. Mead EJ, Maguire JJ, Kuc RE,Davenport AP (2007). Kisspeptins: a multifunctional peptide system with a role in reproduction, cancer and the cardiovascular system. Br J Pharmacol., 151 (8):1143-1153.
| DOI | 名称 | |
|---|---|---|
| 10.1038/s41598-018-20571-2 | Kisspeptin-10 (KP-10) stimulates osteoblast differentiation through GPR54-mediated regulation of BMP2 expression and activation | 下载 |
多肽H2N-Tyr-Asn-Trp-Asn-Ser-Phe-Gly-Leu-Arg-Phe-NH2的合成步骤:
1、合成MBHA树脂:取若干克的MBHA树脂(如初始取代度为0.5mmol/g)和1倍树脂摩尔量的Fmoc-Linker-OH加入到反应器中,加入DMF,搅拌使氨基酸完全溶解。再加入树脂2倍量的DIEPA,搅拌混合均匀。再加入树脂0.95倍量的HBTU,搅拌混合均匀。反应3-4小时后,用DMF洗涤3次。用2倍树脂体积的10%乙酸酐/DMF 进行封端30分钟。然后再用DMF洗涤3次,甲醇洗涤2次,DCM洗涤2次,再用甲醇洗涤2次。真空干燥12小时以上,得到干燥的树脂{Fmoc-Linker-MHBA Resin},测定取代度。这里测得取代度为 0.3mmol/g。结构如下图:

2、脱Fmoc:取2.11g的上述树脂,用DCM或DMF溶胀20分钟。用DMF洗涤2遍。加3倍树脂体积的20%Pip/DMF溶液,鼓氮气30分钟,然后2倍树脂体积的DMF 洗涤5次。得到 H2N-Linker-MBHA Resin 。(此步骤脱除Fmoc基团,茚三酮检测为蓝色,Pip为哌啶)。结构图如下:

3、缩合:取1.9mmol Fmoc-Phe-OH 氨基酸,加入到上述树脂里,加适当DMF溶解氨基酸,再依次加入3.8mmol DIPEA,1.8mmol HBTU。反应30分钟后,取小样洗涤,茚三酮检测为无色。用2倍树脂体积的DMF 洗涤3次树脂。(洗涤树脂,去掉残留溶剂,为下一步反应做准备)。得到Fmoc-Phe-Linker-MBHA Resin。氨基酸:DIPEA:HBTU:树脂=3:6:2.85:1(摩尔比)。结构图如下:

4、依次循环步骤二、步骤三,依次得到
H2N-Phe-Linker-MBHA Resin
Fmoc-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Asn(Trt)-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
H2N-Asn(Trt)-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
Fmoc-Tyr(tBu)-Asn(Trt)-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin
以上中间结构,均可在专肽生物多肽计算器-多肽结构计算器中,一键画出。
最后再经过步骤二得到 H2N-Tyr(tBu)-Asn(Trt)-Trp(Boc)-Asn(Trt)-Ser(tBu)-Phe-Gly-Leu-Arg(Pbf)-Phe-Linker-MBHA Resin,结构如下:

5、切割:6倍树脂体积的切割液(或每1g树脂加8ml左右的切割液),摇床摇晃 2小时,过滤掉树脂,用冰无水乙醚沉淀滤液,并用冰无水乙醚洗涤沉淀物3次,最后将沉淀物放真空干燥釜中,常温干燥24小试,得到粗品H2N-Tyr-Asn-Trp-Asn-Ser-Phe-Gly-Leu-Arg-Phe-NH2。结构图见产品结构图。
切割液选择:1)TFA:H2O=95%:5%
2)TFA:H2O:TIS=95%:2.5%:2.5%
3)三氟乙酸:茴香硫醚:1,2-乙二硫醇:苯酚:水=87.5%:5%:2.5%:2.5%:2.5%
(前两种适合没有容易氧化的氨基酸,例如Trp、Cys、Met。第三种适合几乎所有的序列。)
6、纯化冻干:使用液相色谱纯化,收集目标峰液体,进行冻干,获得蓬松的粉末状固体多肽。不过这时要取小样复测下纯度 是否目标纯度。
7、最后总结:
杭州专肽生物技术有限公司(ALLPEPTIDE https://www.allpeptide.com)主营定制多肽合成业务,提供各类长肽,短肽,环肽,提供各类修饰肽,如:荧光标记修饰(CY3、CY5、CY5.5、CY7、FAM、FITC、Rhodamine B、TAMRA等),功能基团修饰肽(叠氮、炔基、DBCO、DOTA、NOTA等),同位素标记肽(N15、C13),订书肽(Stapled Peptide),脂肪酸修饰肽(Pal、Myr、Ste),磷酸化修饰肽(P-Ser、P-Thr、P-Tyr),环肽(酰胺键环肽、一对或者多对二硫键环),生物素标记肽,PEG修饰肽,甲基化修饰肽
以上所有内容,为专肽生物原创内容,请勿发布到其他网站上。