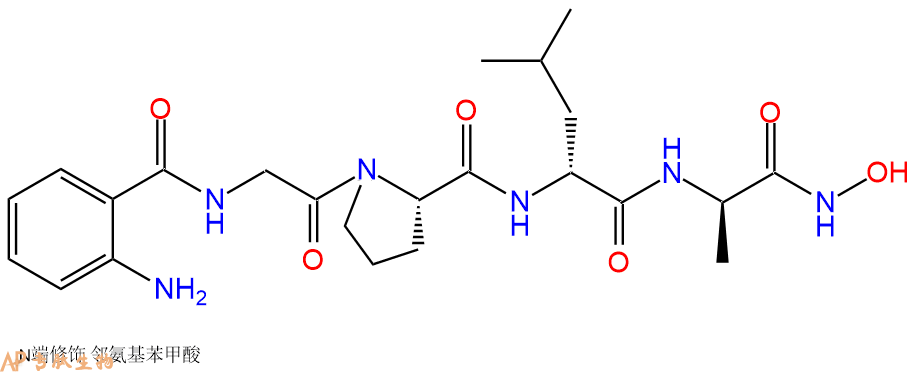
水溶性肽基羟肟酸FN439是一种广谱的人基质金属蛋白酶抑制剂。4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH即使在与pronase或人粒细胞弹性酶长时间孵育后仍保持其活性。FN 439抑制间质和粒细胞胶原酶,粒细胞明胶酶和皮肤成纤维细胞基质酶,IC₅₀分别为1·10⁻⁶M, 3·10⁻⁴M和1.5·10⁻⁴M。
编号:431220
CAS号:124168-73-6
单字母:Abz-GPla-NHOH
| 编号: | 431220 |
| 中文名称: | FN-439, MMP Inhibitor 1 |
| 英文名: | 4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH trifluoroacetate salt |
| 英文同义词: | 4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH; 4-aminobenzoyl-glycyl-prolyl-leucyl-alanine hydroxamic acid; MMP Inhibitor I |
| CAS号: | 124168-73-6 |
| 单字母: | Abz-GPla-NHOH |
| 三字母: | Abz N端Abz修饰 -Gly甘氨酸 -Pro脯氨酸 -DLeuD型亮氨酸 -DAlaD型丙氨酸 -NHOH暂无说明 |
| 氨基酸个数: | 4 |
| 分子式: | C23H34N6O6 |
| 平均分子量: | 490.55 |
| 精确分子量: | 490.25 |
| 等电点(PI): | - |
| pH=7.0时的净电荷数: | - |
| 平均亲水性: | -1.15 |
| 疏水性值: | 0.9 |
| 消光系数: | - |
| 标签: | D型氨基酸肽 抑制剂相关肽(Inhibitor Peptide) |
The water-soluble peptidylhydroxamic acid FN439 is a broad-spectrum inhibitor of human matrix metalloproteinases. 4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH retains its activity even after prolonged incubation with pronase or human granulocyte elastase. FN 439 inhibits interstitial and granulocyte collagenases, granulocyte gelatinase and skin fibroblast stromelysin with IC₅₀ of 1 · 10⁻⁶ M, 3 · 10⁻⁵ M and 1.5 · 10⁻⁴ M, respectively.
水溶性肽基羟肟酸FN439是一种广谱的人基质金属蛋白酶抑制剂。4-Abz-Gly-Pro-D-Leu-D-Ala-NHOH即使在与pronase或人粒细胞弹性酶长时间孵育后仍保持其活性。FN 439抑制间质和粒细胞胶原酶,粒细胞明胶酶和皮肤成纤维细胞基质酶,IC₅₀分别为1·10⁻⁶M, 3·10⁻⁴M和1.5·10⁻⁴M。
很多蛋白在细胞中非常容易被降解,或被标记,进而被选择性地破坏。但含有部分D型氨基酸的多肽则显示了很强的抵抗蛋白酶降解能力。
定义
酶是用于生化反应的非常有效的催化剂。它们通过提供较低活化能的替代反应途径来加快反应速度。酶作用于底物并产生产物。一些物质降低或什至停止酶的催化活性被称为抑制剂。
发现
1965年,Umezawa H分析了微生物产生的酶抑制剂,并分离出了抑制亮肽素和抗痛药的胰蛋白酶和木瓜蛋白酶,乳糜蛋白酶抑制的胰凝乳蛋白酶,胃蛋白酶抑制素抑制胃蛋白酶,泛磷酰胺抑制唾液酸酶,乌藤酮抑制酪氨酸羟化酶,多巴汀抑制多巴胺3-羟硫基嘧啶和多巴胺3-羟色胺酶酪氨酸羟化酶和多巴胺J3-羟化酶。最近,一种替代方法已应用于预测新的抑制剂:合理的药物设计使用酶活性位点的三维结构来预测哪些分子可能是抑制剂1。已经开发了用于识别酶抑制剂的基于计算机的方法,例如分子力学和分子对接。
结构特征
已经确定了许多抑制剂的晶体结构。已经确定了三种与凝血酶复合的高效且选择性的低分子量刚性肽醛醛抑制剂的晶体结构。这三种抑制剂全部在P3位置具有一个新的内酰胺部分,而对胰蛋白酶选择性最高的两种抑制剂在P1位置具有一个与S1特异性位点结合的胍基哌啶基。凝血酶的抑制动力学从慢到快变化,而对于胰蛋白酶,抑制的动力学在所有情况下都快。根据两步机理2中稳定过渡态络合物的缓慢形成来检验动力学。
埃米尔•菲舍尔(Emil Fischer)在1894年提出,酶和底物都具有特定的互补几何形状,彼此恰好契合。这称为“锁和钥匙”模型3。丹尼尔·科什兰(Daniel Koshland)提出了诱导拟合模型,其中底物和酶是相当灵活的结构,当底物与酶4相互作用时,活性位点通过与底物的相互作用不断重塑。
在众多生物活性肽的成熟过程中,需要由其谷氨酰胺(或谷氨酰胺)前体形成N末端焦谷氨酸(pGlu)。游离形式并与底物和三种咪唑衍生抑制剂结合的人QC的结构揭示了类似于两个锌外肽酶的α/β支架,但有多个插入和缺失,特别是在活性位点区域。几种活性位点突变酶的结构分析为针对QC相关疾病5的抑制剂的合理设计提供了结构基础。
作用方式
酶是催化化学反应的蛋白质。酶与底物相互作用并将其转化为产物。抑制剂的结合可以阻止底物进入酶的活性位点和/或阻止酶催化其反应。抑制剂的种类繁多,包括:非特异性,不可逆,可逆-竞争性和非竞争性。可逆抑制剂 以非共价相互作用(例如疏水相互作用,氢键和离子键)与酶结合。非特异性抑制方法包括最终使酶的蛋白质部分变性并因此不可逆的任何物理或化学变化。特定抑制剂 对单一酶发挥作用。大多数毒药通过特异性抑制酶发挥作用。竞争性抑制剂是任何与底物的化学结构和分子几何结构非常相似的化合物。抑制剂可以在活性位点与酶相互作用,但是没有反应发生。非竞争性抑制剂是与酶相互作用但通常不在活性位点相互作用的物质。非竞争性抑制剂的净作用是改变酶的形状,从而改变活性位点,从而使底物不再能与酶相互作用而产生反应。非竞争性抑制剂通常是可逆的。不可逆抑制剂与酶形成牢固的共价键。这些抑制剂可以在活性位点附近或附近起作用。
功能
工业应用中, 酶在商业上被广泛使用,例如在洗涤剂,食品和酿造工业中。蛋白酶用于“生物”洗衣粉中,以加速蛋白质在诸如血液和鸡蛋等污渍中的分解。商业上使用酶的问题包括:它们是水溶性的,这使得它们难以回收,并且一些产物可以抑制酶的活性(反馈抑制)。
药物分子,许多药物分子都是酶抑制剂,药用酶抑制剂通常以其特异性和效力为特征。高度的特异性和效力表明该药物具有较少的副作用和较低的毒性。酶抑制剂在自然界中发现,并且也作为药理学和生物化学的一部分进行设计和生产6。
天然毒物 通常是酶抑制剂,已进化为保护植物或动物免受天敌的侵害。这些天然毒素包括一些已知最剧毒的化合物。
神经气体( 例如二异丙基氟磷酸酯(DFP))通过与丝氨酸的羟基反应生成酯,从而抑制了乙酰胆碱酯酶的活性位点。
参考
1、Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
2、Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
3、Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
4、Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
5、Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
6、Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
Definition
Enzymes are very efficient catalysts for biochemical reactions. They speed up reactions by providing an alternative reaction pathway of lower activation energy. Enzyme acts on substrate and gives rise to a product. Some substances reduce or even stop the catalytic activities of enzymes are called inhibitors.
Discovery
In 1965, Umezawa H analysed enzyme inhibitors produced by microorganisms and isolated leupeptin and antipain inhibiting trypsin and papain, chymostatin inhibiting chymotrypsin, pepstatin inhibiting pepsin, panosialin inhibiting sialidases, oudenone inhibiting tyrosine hydroxylase, dopastin inhibiting dopamine 3-hydroxylase, aquayamycin and chrothiomycin inhibiting tyrosine hydroxylase and dopamine J3-hydroxylase . Recently, an alternative approach has been applied to predict new inhibitors: rational drug design uses the three-dimensional structure of an enzyme's active site to predict which molecules might be inhibitors 1. Computer-based methods for identifying inhibitor for an enzyme have been developed, such as molecular mechanics and molecular docking.
Structural Characteristics
The crystal structures of many inhibitors have been determined. The crystal structures of three highly potent and selective low-molecular weight rigid peptidyl aldehyde inhibitors complexed with thrombin have been determined. All the three inhibitors have a novel lactam moiety at the P3 position, while the two with greatest trypsin selectivity have a guanidinopiperidyl group at the P1 position that binds in the S1 specificity site. The kinetics of inhibition vary from slow to fast with thrombin and are fast in all cases with trypsin. The kinetics are examined in terms of the slow formation of a stable transition-state complex in a two-step mechanism 2.
Emil Fischer in 1894 suggested that both the enzyme and the substrate possess specific complementary geometric shapes that fit exactly into one another.This is known as "the lock and key" model 3. Daniel Koshland suggested induced fit model where substrate and enzymes are rather flexible structures, the active site is continually reshaped by interactions with the substrate as the substrate interacts with the enzyme 4.
N-terminal pyroglutamate (pGlu) formation from its glutaminyl (or glutamyl) precursor is required in the maturation of numerous bioactive peptides. The structure of human QC in free form and bound to a substrate and three imidazole-derived inhibitors reveals an alpha/beta scaffold akin to that of two-zinc exopeptidases but with several insertions and deletions, particularly in the active-site region. The structural analyses of several active-site-mutant enzymes provide a structural basis for the rational design of inhibitors against QC-associated disorders 5.
Mode of Action
Enzymes are proteins that catalyze chemical reactions. Enzymes interact with substrate and convert them into products. Inhibitor binding can stop a substrate from entering the enzyme's active site and/or hinder the enzyme from catalyzing its reaction. There are a variety of types of inhibitors including: nonspecific, irreversible, reversible - competitive and noncompetitive. Reversible inhibitors bind to enzymes with non-covalent interactions like hydrophobic interactions, hydrogen bonds, and ionic bonds. Non-specific methods of inhibition include any physical or chemical changes which ultimately denature the protein portion of the enzyme and are therefore irreversible. Specific Inhibitors exert their effects upon a single enzyme. Most poisons work by specific inhibition of enzymes. A competitive inhibitor is any compound which closely resembles the chemical structure and molecular geometry of the substrate. The inhibitor may interact with the enzyme at the active site, but no reaction takes place. A noncompetitive inhibitor is a substance that interacts with the enzyme, but usually not at the active site. The net effect of a non competitive inhibitor is to change the shape of the enzyme and thus the active site, so that the substrate can no longer interact with the enzyme to give a reaction. Non competitive inhibitors are usually reversible. Irreversible Inhibitors form strong covalent bonds with an enzyme. These inhibitors may act at, near, or remote from the active site .
Functions
Industrial application, enzymes are widely used commercially, for example in the detergent, food and brewing industries. Protease enzymes are used in 'biological' washing powders to speed up the breakdown of proteins in stains like blood and egg. Problems using enzymes commercially include: they are water soluble which makes them hard to recover and some products can inhibit the enzyme activity (feedback inhibition) .
Drug molecules, many drug molecules are enzyme inhibitors and a medicinal enzyme inhibitor is usually characterized by its specificity and its potency. A high specificity and potency suggests that a drug will have fewer side effects and less toxic. Enzyme inhibitors are found in nature and are also designed and produced as part of pharmacology and biochemistry 6.
Natural poisons are often enzyme inhibitors that have evolved to defend a plant or animal against predators. These natural toxins include some of the most poisonous compounds known.
Nerve gases such as diisopropylfluorophosphate (DFP) inhibit the active site of acetylcholine esterase by reacting with the hydroxyl group of serine to make an ester.
References
Scapin G (2006). Structural biology and drug discovery. Curr. Pharm. Des., 12(17):2087–2097.
Krishnan R, Zhang E, Hakansson K, Arni RK, Tulinsky A, Lim-Wilby MS, Levy OE, Semple JE, Brunck TK (1998). Highly selective mechanism-based thrombin inhibitors: structures of thrombin and trypsin inhibited with rigid peptidyl aldehydes. Biochemistry, 37 (35):12094-12103.
Fischer E (1894). Einfluss der configuration auf die wirkung der enzyme. Ber. Dt. Chem. Ges., 27:2985–2993.
Koshland DE (1958). Application of a theory of enzyme specificity to protein synthesis. PNAS., 44 (2):98–104.
Huang KF, Liu YL, Cheng WJ, Ko TP, Wang AH (2005). Crystal structures of human glutaminyl cyclase, an enzyme responsible for protein N-terminal pyroglutamate formation. PNAS., 102(37):13117-13122.
Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN (2002). Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Curr Med Chem., 9(22):1981-1989.
1.Synergistic Effect of the Combination of Novel Suberoylanilide Hydroxamic Acid Derivatives with Cisplatin on Anti-proliferation of Human Cancer Cells.
Xie R, Shi J, Cheng C, Yun F, Liu X, Tang P, Wu X, Yang M, Yuan Q1. Med Chem. 2016 Apr 4. [Epub ahead of print]
A novel, green, and atom-economical boric acid catalyzed direct amidation without the use of any coupling agents for the preparation of suberoylanilide hydroxamic acid (SAHA) and SAHA-based inhibitors targeting anti-proliferation of cancer cells is provided. The new SAHA-based inhibitor B123, when used alone, exhibited higher anti-proliferative activities than SAHA or Cisplatin against a number of human cancer cells. We have examined the effect of combination of these SAHA-based inhibitors with Cisplatin. We found synergistic effects of the combination of SAHA-based inhibitors with Cisplatin over a wide range of concentrations against human liver cancer cells HepG2 and two human lung cancer cell lines H1299 and H460. This synergism leads to up to 8-fold of dose reduction for Cisplatin in the combination with our synthesized inhibitor B123 against H1299.
2.Disarming an Electrophilic Warhead: Retaining Potency in Tyrosine Kinase Inhibitor (TKI)-Resistant CML Lines While Circumventing Pharmacokinetic Liabilities.
Ali AM1,2, Gómez-Biagi RF1, Rosa DA1, Lai PS1, Heaton WL3, Park JS1, Eiring AM4, Vellore NA4, de Araujo ED1, Ball DP1, Shouksmith AE1, Patel AB4, Deininger MW4, O'Hare T4, Gunning PT5. ChemMedChem. 2016 Mar 30. doi: 10.1002/cmdc.201600021. [Epub ahead of print]
Pharmacologic blockade of the activation of signal transducer and activator of transcription 3 (STAT3) in tyrosine kinase inhibitor (TKI)-resistant chronic myeloid leukemia (CML) cell lines characterized by kinase-independent resistance was shown to re-sensitize CML cells to TKI therapy, suggesting that STAT3 inhibitors in combination with TKIs are an effective combinatorial therapeutic for the treatment of CML. Benzoic acid- and hydroxamic acid-based STAT3 inhibitors SH-4-054 and SH-5-007, developed previously in our laboratory, demonstrated promising activity against these resistant CML cell lines. However, pharmacokinetic studies in murine models (CD-1 mice) revealed that both SH-4-054 and SH-5-007 are susceptible to glutathione conjugation at the para position of the pentafluorophenyl group via nucleophilic aromatic substitution (SN Ar). To determine whether the electrophilicity of the pentafluorophenyl sulfonamide could be tempered, an in-depth structure-activity relationship (SAR) study of the SH-4-054 scaffold was conducted.
3.Unprecedented binding mode of hydroxamate-based inhibitors of glutamate carboxypeptidase II: structural characterization and biological activity.
Novakova Z, Wozniak K, Jancarik A, Rais R, Wu Y, Pavlicek J, Ferraris DV, Havlinova B, Ptacek J, Vavra J, Hin N, Rojas C, Majer P, Slusher BS, Tsukamoto T, Barinka C. J Med Chem. 2016 Apr 13. [Epub ahead of print]
Inhibition of glutamate carboxypeptidase II (GCPII) is effective in preclinical models of neurological disorders associated with excessive activation of glutamatergic systems. Here we report synthesis, structural characterization and biological activity of new hydroxamic acid-based inhibitors with nanomolar affinity for human GCPII. Crystal structures of GCPII/hydroxamate complexes revealed an unprecedented binding mode in which the putative P1' glutarate occupies the spacious entrance funnel, rather than the conserved glutamate-binding S1' pocket. This unique binding mode provides a mechanistic explanation for the structure-activity relationship data, most notably the lack of enantiospecificity and the tolerance for bulky/hydrophobic functions as substituents of a canonical glutarate moiety. The in vivo pharmacokinetics profile of one of the inhibitors will be presented along with analgesic efficacy data from the rat chronic constrictive injury model of neuropathic pain.
4.Histone deacetylase inhibitor-induced cancer stem cells exhibit high pentose phosphate pathway metabolism.
Debeb BG1,2, Lacerda L1,2, Larson R1,2, Wolfe AR1,2, Krishnamurthy S3,2, Reuben JM4,2, Ueno NT5,2, Gilcrease M3, Woodward WA1,2. Oncotarget. 2016 Apr 7. doi: 10.18632/oncotarget.8631. [Epub ahead of print]
PURPOSE: We recently demonstrated that histone deacetylase (HDAC) inhibitors can "reprogram" differentiated triple-negative breast cancer cells to become quiescent stem-like cancer cells. We hypothesized that the metabolic state of such cells differs from that of their differentiated progeny.